
Cross-Assembly of Metagenomes | Help page
Metagenomes are often characterized by high levels of unknown reads with no similarity to any sequences in Genbank. Although these are often discarded from analysis, they contain a wealth of information for comparative metagenomics.
crAss is a tool that enables fast and intuitive analysis of complete metagenomic data sets by counting the number of shared contigs between samples in a cross-assembly of all reads.
Prior to running crAss, you will need to combine all your metagenomic data sets into a single cross-assembly using your favorite de novo assembly tool. Note that the read identifiers need to be unique across all the data set files, otherwise crAss will not be able to recognize to which data set a read belongs when reading the cross-assembly ACE file.
The input to the web server includes the cross-assembly ACE file and the individual read files in FASTA or FASTQ format. However, not all data in these files is used by the crAss program. To quicken the upload process, you can select the relevant parts of the file for crAss to upload using the commands below. As this removes the bulk of the file (the sequences), it can lead to a tremendous decrease in the file size, saving you time and us disk space! Also, the above approach is useful if you do not want to upload your sequences to our server, e.g. because they are private.
The command to decrease the size of the ACE file:
grep -E '^CO|AF' bigFile.ace > smallFile.ace
The command to decrease the size of the FASTA files:
grep '>' bigFile.fasta > smallFile.fasta
Upload the files by clicking "Add ACE / FASTA file" and selecting the appropriate files on your computer.

Note that the input files can be zipped (using GZIP or ZIP) which might be useful if you have a slow internet connection, but the original file extensions (for example .fasta or .ace) have to be included in the file name.
To start the upload, click "Start upload" or the arrow button.
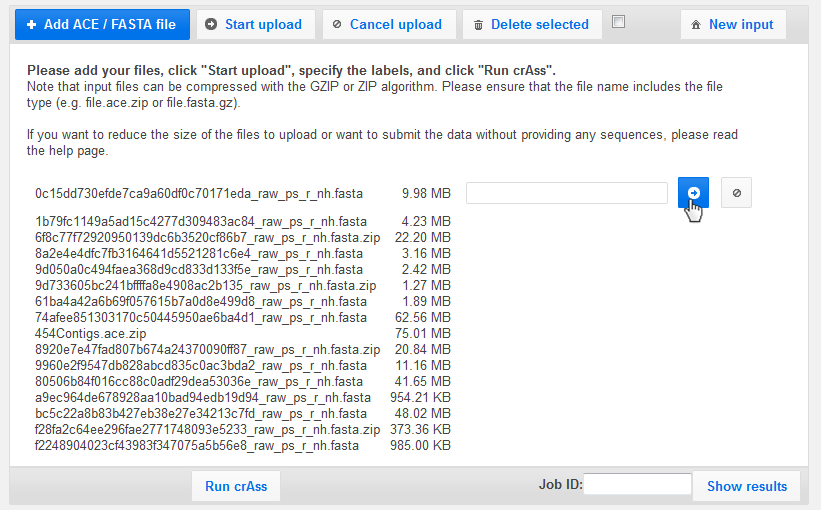
The progress bar indicates that the files are uploading...
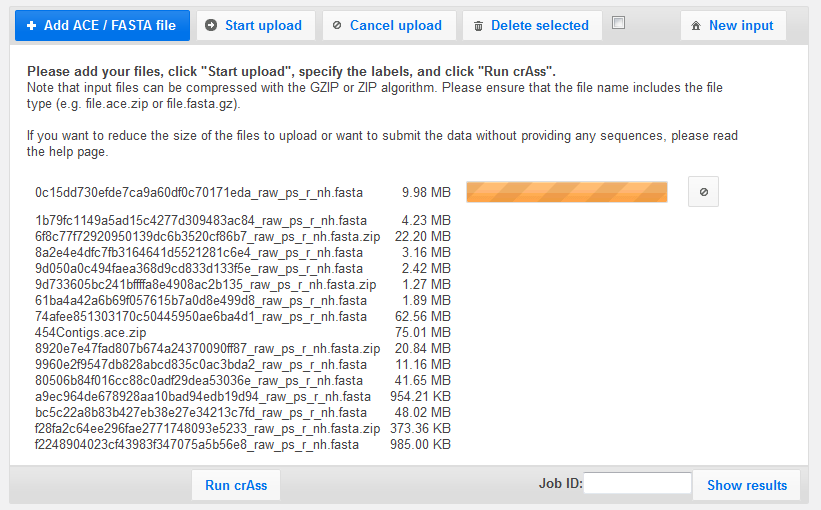
On the next screen, you have the option of changing the names of the files for each data set.
It is important that every data set has a unique name!
Note that the name of the ACE file does not need to be changed, as it is not one of the primary data sets.
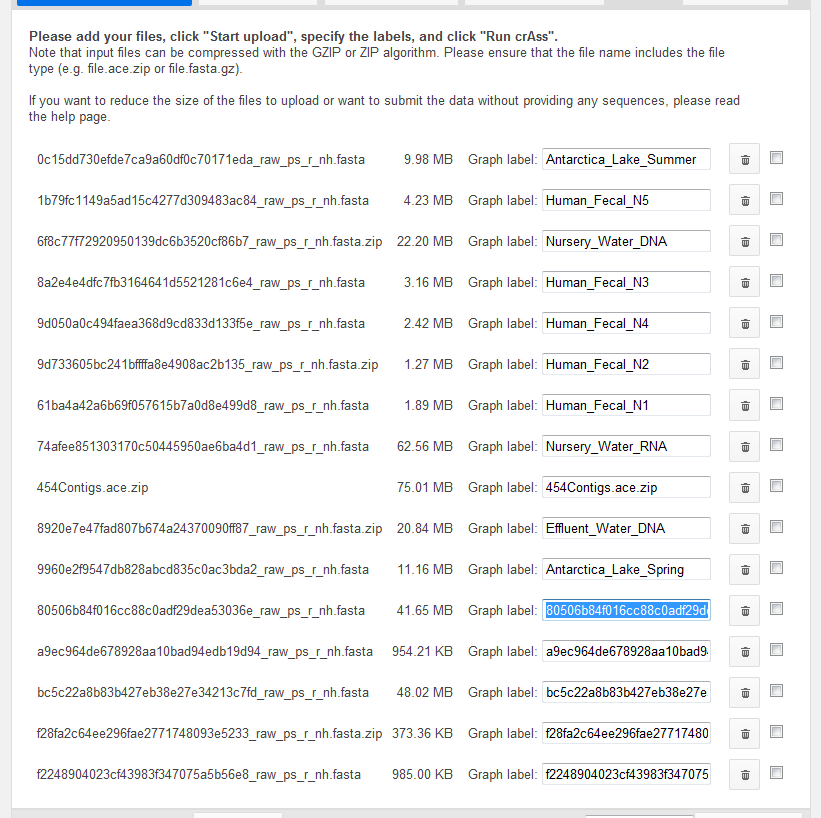
Deleting a file is easy with the trash button, or by clicking the checkboxes and clicking "Delete selected" at the top of the frame.

Start the process by clicking "Run crAss" at the bottom.
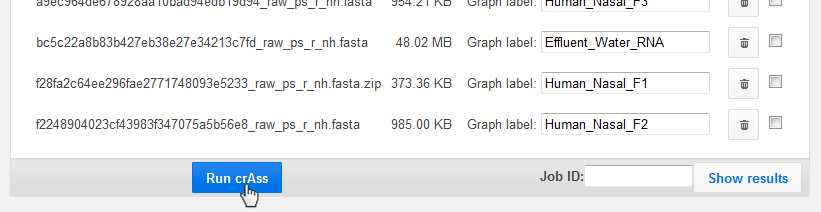
You may need to wait until your zipped files have been extracted.
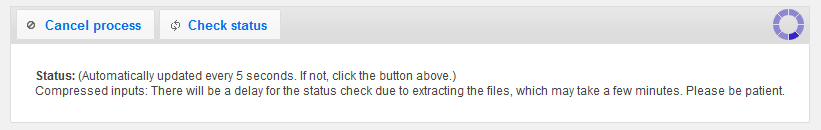
While crAss is running, the page will refresh automatically until the process is done.
Ususally, this will take less than a minute.
During this time, crAss extracts all contigs, calculates pair-wise distances between the metagenomes and builds an output image to visualize your data.
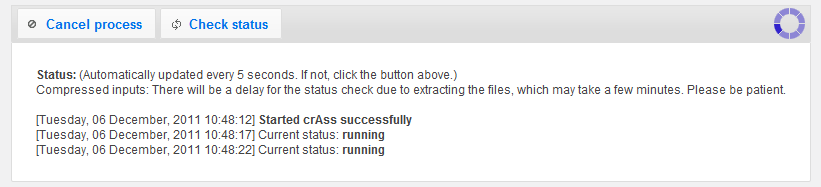
The first thing to notice after the run is done, is your Job ID.

With this Job ID, you can always recover your results at a later time just by entering it on the crAss home page.
Just enter it in the "Job ID" field and click "Show results".

You can download the names you gave to each of the data sets for future reference.

The first output file is a file that contains a single line for each contig and shows how many reads from each data set it contains.
This file also lists the unassembled reads.
Download this file if you want to further analyze the read distribution across cross-contigs.

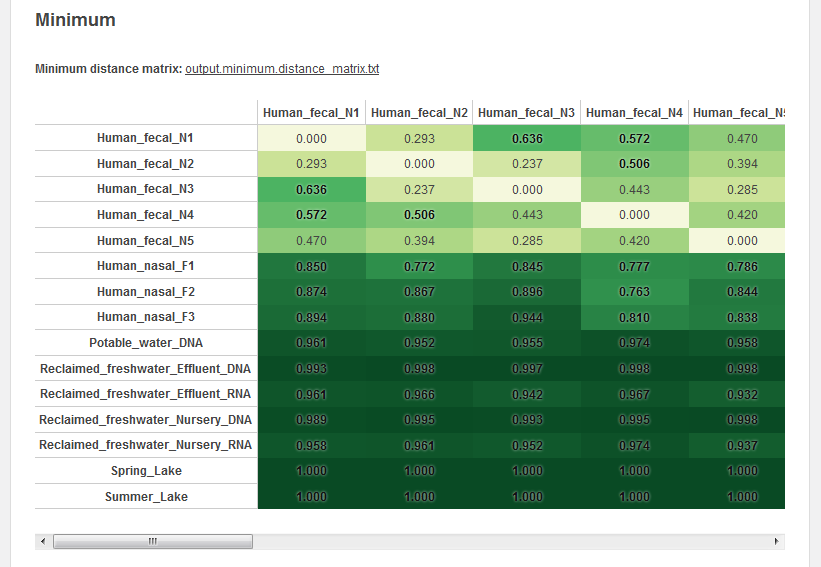
The second output of crAss is series of symmetrical distance matrices.
We construct these distance matrices according to different distance formulas, constructed as explained in our paper.
The matrices are visualized on the output page and can be downloaded as a .txt file.
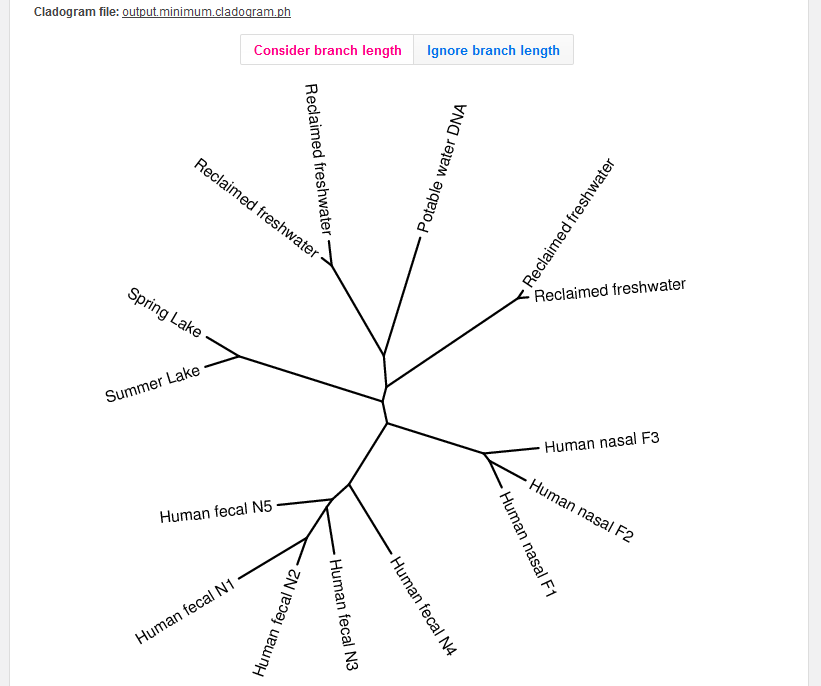
The third output is an image that displays the similarities between metagenomes.
If you included 4 or more data sets in the cross-assembly, the output image will be a cladogram built from the distance matrix using BioNJ (Gascuel, 1997).
As this cladogram is based on the distance matrix, we construct separate cladograms for each distance formula.
You can choose to display or ignore the branch lengths.
You can download the Phylip bracketnotation (the "output.XXX.cladogram.ph" file) for use in other programs.
If you included 2 or 3 data sets in the cross-assembly, a cladogram is not meaningful so the output image will be a 2D or 3D graph.
Each dot represents a contig, and the X/Y/Z coordinates show how many reads from each data set were included in that contig.
These 2D and 3D graphs are based on the data in "output.contigs2reads.txt", so you can always download that file for personalized visualization of the results in another program.
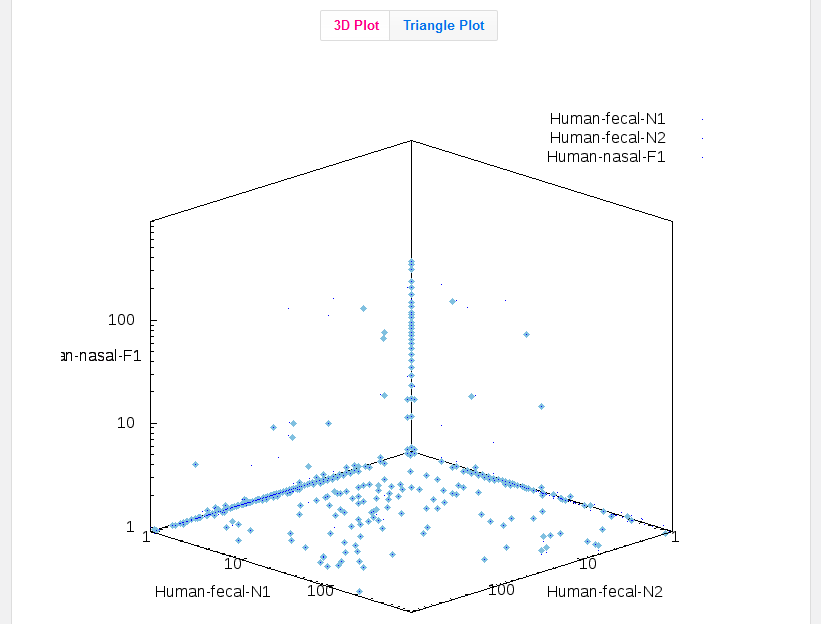
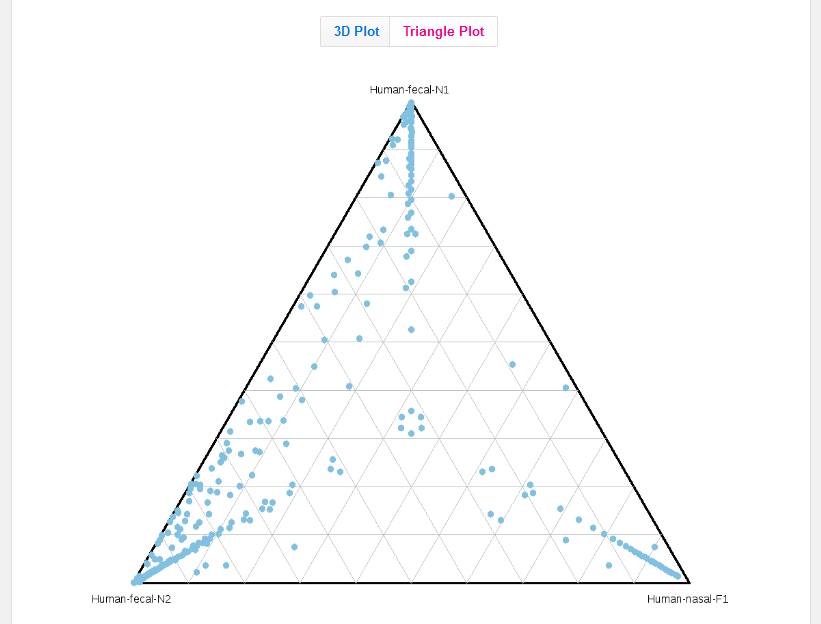
3D graphs are also represented as a triangle plot.
If you use crAss in your research, please cite:
Bas E. Dutilh, Robert Schmieder, Jim Nulton, Ben Felts, Peter Salamon, Forest Rohwer, Robert A. Edwards and John L. Mokili,
"Reference-independent comparative metagenomics using cross-assembly: crAss", Bioinformatics. Pubmed, PDF.
Website: http://edwards.sdsu.edu/crass
Download: http://sourceforge.net/p/crass
Forum: http://seqanswers.com/forums/showthread.php?p=65129
Contact: http://www.cmbi.ru.nl/~dutilh/